


What are the primary symptoms of thalassemia?
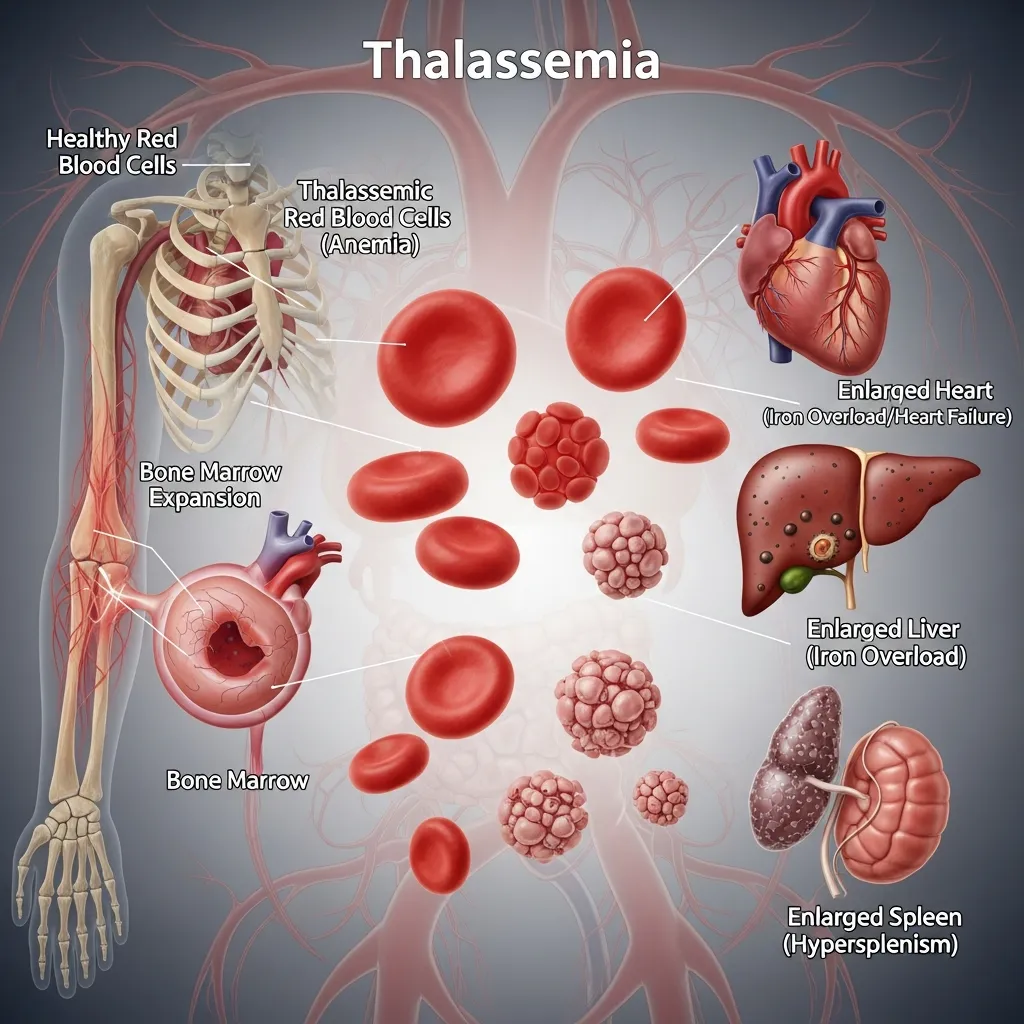
The symptoms of thalassemia vary widely depending on the number of mutated or deleted genes and the overall severity of the condition. Mild forms often present no outward signs and may go unnoticed for years, with individuals only discovering the condition during routine blood tests. In moderate to severe cases, symptoms are more noticeable and can significantly affect daily life.
Common signs include chronic fatigue, weakness, dizziness, and pale or yellowish skin due to reduced oxygen delivery in the blood. Some individuals may also experience dark urine, irritability, and shortness of breath, especially during physical activity. As the body attempts to compensate for low red blood cell levels, the bone marrow becomes overactive, which can lead to bone expansion and, in severe cases, facial bone deformities.
Children with more severe forms of thalassemia may show additional symptoms such as delayed growth, poor appetite, and delayed puberty. Over time, repeated anemia can also affect heart function and overall physical development if not properly managed. These symptoms highlight the importance of early recognition and ongoing medical care.
What diagnostic procedures confirm a thalassemia diagnosis?
Accurate diagnosis of thalassemia begins with a Complete Blood Count (CBC), which measures red blood cell size, hemoglobin levels, and overall blood health. One of the key indicators is a low Mean Corpuscular Volume (MCV), which suggests smaller-than-normal red blood cells. However, CBC alone is not enough to confirm the condition.
To further investigate, healthcare providers use hemoglobin electrophoresis, a test that separates and measures different types of hemoglobin in the blood. This helps identify abnormal hemoglobin patterns associated with beta thalassemia and other related disorders. In some cases, additional iron studies are performed to rule out iron deficiency anemia, which can present with similar symptoms.
The most definitive method for diagnosis is targeted genetic DNA testing. This test identifies specific mutations or deletions in the alpha or beta globin genes, confirming not only the presence of thalassemia but also its exact type. Genetic testing is especially important for carrier detection and family planning, as it provides precise information about inheritance risk.
Together, these diagnostic procedures allow doctors to accurately classify the condition, determine its severity, and develop an appropriate treatment plan tailored to the patient’s needs.
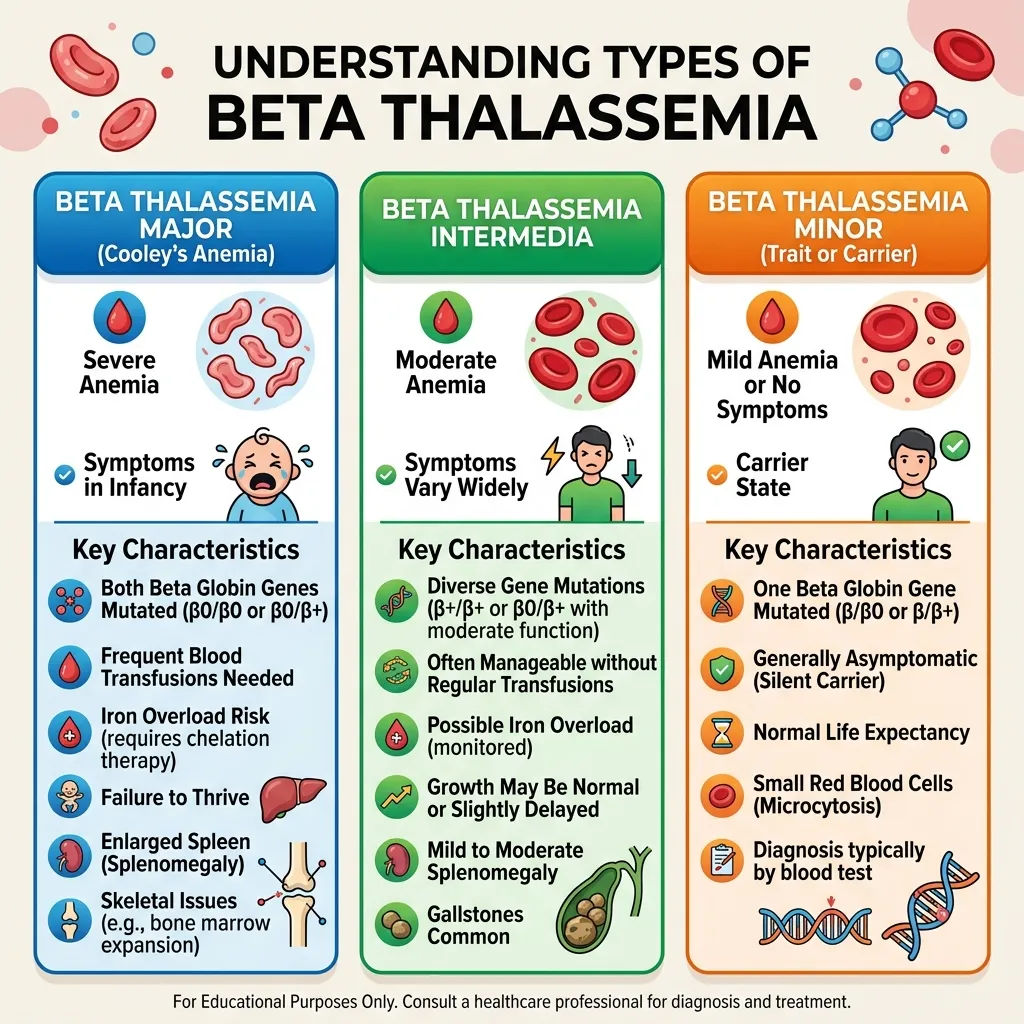
 Treatment plans for thalassemia are closely aligned with the severity of the diagnosis and the specific type of the disorder. Individuals with thalassemia trait or minor forms typically do not require active medical treatment, although regular monitoring may be recommended to track hemoglobin levels and avoid misdiagnosis as iron deficiency anemia. In some cases, patients may only need dietary guidance and periodic blood tests to ensure overall stability.
Treatment plans for thalassemia are closely aligned with the severity of the diagnosis and the specific type of the disorder. Individuals with thalassemia trait or minor forms typically do not require active medical treatment, although regular monitoring may be recommended to track hemoglobin levels and avoid misdiagnosis as iron deficiency anemia. In some cases, patients may only need dietary guidance and periodic blood tests to ensure overall stability.


Frequently Asked Questions (FAQs)
1. What is the main focus of thalassemia awareness?
Thalassemia awareness focuses on educating the public about this inherited blood disorder, promoting early genetic screening, and providing accurate medical information to help individuals manage their health effectively.
It also helps reduce stigma and encourages families to seek timely medical advice before complications develop.
2. Is thalassemia contagious?
No. Thalassemia is a strictly genetic condition inherited from parents. You cannot catch it from another person through contact or environmental exposure.
It is passed through genes, which makes family history and screening very important.
3. Do all thalassemia patients require regular blood transfusions?
No. Only individuals with severe forms, such as beta thalassemia major or Hemoglobin H disease, require regular transfusions. Carriers and those with minor traits typically do not need treatment.
Mild cases often live normal lives with only routine monitoring and no active intervention.
4. Can a carrier of thalassemia develop the severe form of the disease later in life?
No. Your genetic makeup does not change over time. If you are diagnosed as a carrier or with a minor trait, it will not progress into a severe disease.
However, awareness of carrier status remains important for future family planning decisions.
5. Why should thalassemia patients avoid iron supplements?
Patients with thalassemia often absorb iron more readily. Taking unnecessary iron supplements can lead to toxic iron overload, which damages the heart, liver, and endocrine system.
Iron should only be taken if a confirmed deficiency is diagnosed by a healthcare professional.
6. What role does folic acid play in thalassemia management?
Folic acid provides essential building blocks that the bone marrow uses to produce new red blood cells, helping the body manage the high cell turnover associated with chronic anemia.
It is commonly recommended as a supportive supplement in many treatment plans.
7. Can people with thalassemia exercise safely?
Yes. Mild to moderate, low-impact exercise is encouraged to support cardiovascular health. However, individuals with severe forms or enlarged spleens should consult their doctor before engaging in high-impact or contact sports.
Regular physical activity can also help improve energy levels and overall well-being.
8. How is thalassemia differentiated from iron deficiency anemia?
While both conditions cause small, pale red blood cells, doctors differentiate them using iron studies (ferritin levels) and genetic DNA testing or hemoglobin electrophoresis.
Accurate diagnosis is essential to avoid incorrect treatment and unnecessary iron supplementation.
9. What should couples do if they both carry a thalassemia trait?
Couples should seek genetic counseling before pregnancy to understand their risks. Counselors can outline options like prenatal testing or preimplantation genetic diagnosis.
This helps families make informed decisions and reduce the risk of severe thalassemia in children.
10. Is there a permanent cure for thalassemia?
Currently, bone marrow or stem cell transplantation is the only potential cure for severe thalassemia. Gene therapy and CRISPR technologies are in clinical trials and show strong promise for the future.
Ongoing research continues to improve safety and accessibility of these advanced treatments.