


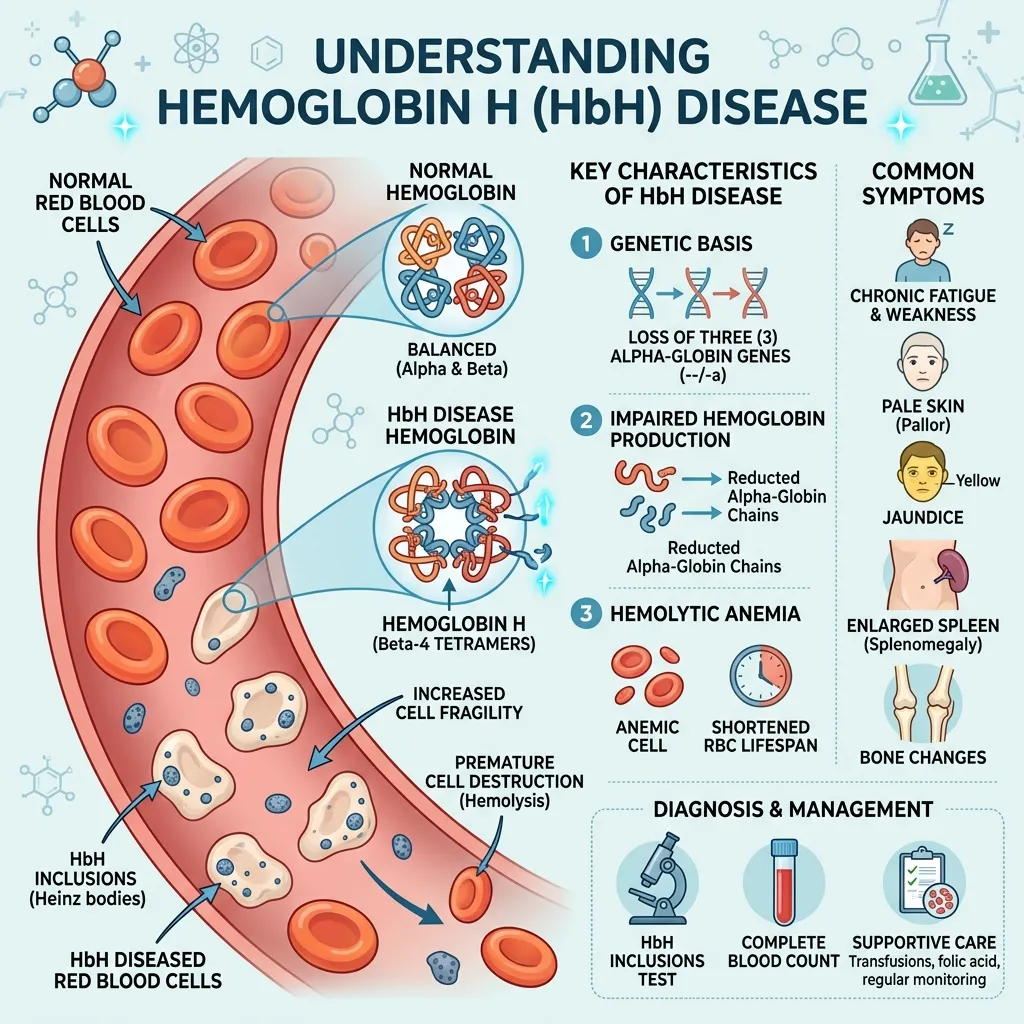
The condition develops from a genetic problem affecting the alpha-globin genes. Specifically, three of the four alpha-globin genes are missing or faulty. This leaves the body struggling to make enough normal hemoglobin, leading to chronic anemia and a cascade of related signs and symptoms.
Understanding HbH disease symptoms is important for one clear reason: early recognition. Because the condition is lifelong, knowing what to expect helps patients and families work with doctors to manage symptoms, avoid complications, and maintain a good quality of life. The earlier alpha thalassemia HbH disease is identified, the sooner proper care can begin.
The Genetic Basis of HbH Disease
To understand HbH disease symptoms, it helps to start with the genetics behind the condition. Alpha thalassemia HbH disease is rooted in how four specific genes behave.
Deletion of Three Alpha-Globin Genes
Every person normally carries four alpha-globin genes—two inherited from each parent. These genes, located on chromosome 16, produce alpha-globin, an essential building block of hemoglobin. In Hemoglobin H disease, three of these four genes are deleted or non-functional. Only one working gene remains.
With just a single functioning gene, the body produces far too little alpha-globin. The result is a serious imbalance that triggers the chronic anemia central to HbH disease.
How This Leads to the Formation of Hemoglobin H
When alpha-globin runs short, the excess beta-globin chains have nothing to pair with. Instead, four beta-globin chains clump together to form an abnormal hemoglobin known as Hemoglobin H, or HbH. This unstable hemoglobin gives the disease its name.
The trouble is that HbH doesn’t carry oxygen efficiently, and it damages red blood cells from the inside. These faulty cells break down faster than normal—a process called hemolysis—which feeds directly into many of the disease’s symptoms.
How HbH Disease Differs from Other Forms of Alpha Thalassemia
The number of affected genes defines where a person falls on the alpha thalassemia spectrum. A silent carrier has one missing gene and no symptoms. The alpha thalassemia trait involves two missing genes and usually causes mild or no symptoms—you can read more in this guide on alpha thalassemia trait symptoms. HbH disease, with three missing genes, sits a step further along, producing moderate anemia. The most severe form, hydrops fetalis, involves all four missing genes and is usually fatal.
This distinction matters. HbH disease causes noticeable symptoms that the milder forms do not, which is why accurate diagnosis is so valuable.
Core HbH Disease Symptoms: A Comprehensive Overview
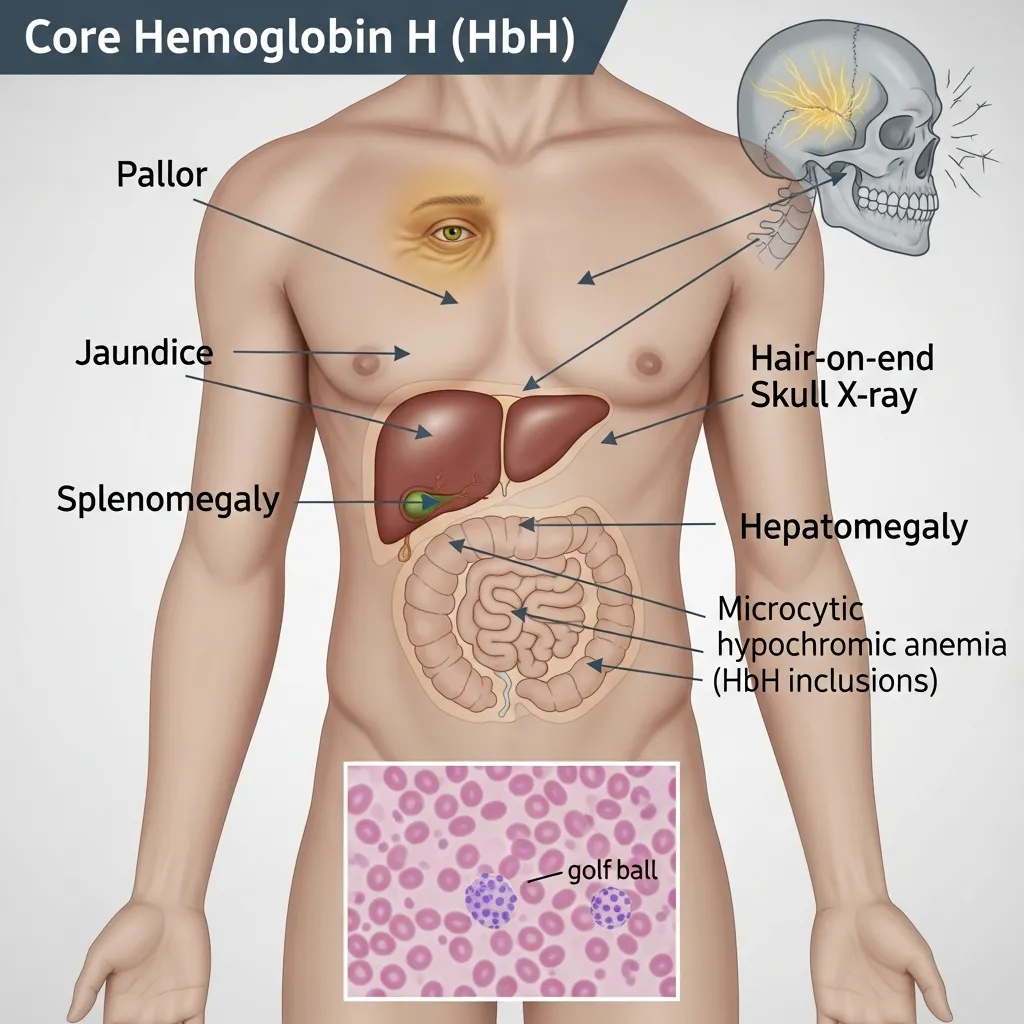 The signs and symptoms of Hemoglobin H disease stem from one central problem—chronic anemia driven by the rapid breakdown of unstable red blood cells. Below are the most common HbH disease signs and symptoms, explained one by one.
The signs and symptoms of Hemoglobin H disease stem from one central problem—chronic anemia driven by the rapid breakdown of unstable red blood cells. Below are the most common HbH disease signs and symptoms, explained one by one.
Chronic Anemia
Chronic anemia is the hallmark of Hemoglobin H disease symptoms. Because the body can’t produce enough healthy hemoglobin, oxygen delivery to tissues drops. Two processes drive this anemia: ineffective erythropoiesis (the bone marrow producing faulty red blood cells) and hemolysis (those cells breaking down too quickly).
The everyday effects of chronic anemia include:
- Fatigue: A persistent, deep tiredness that rest doesn’t fully fix.
- Weakness: Reduced strength and stamina during normal activities.
- Pallor: Pale skin, caused by lower hemoglobin levels.
- Shortness of breath: Especially during physical activity, as the body struggles to deliver enough oxygen.
These symptoms tend to be more pronounced than in the alpha thalassemia trait, where most carriers feel completely healthy.
Splenomegaly (Enlarged Spleen)
The spleen acts as a filter, removing damaged red blood cells from circulation. In Hemoglobin H disease, the spleen works overtime to clear the constant supply of faulty cells. Over time, this extra workload causes it to enlarge—a condition called splenomegaly.
An enlarged spleen can cause abdominal discomfort or a feeling of fullness. It may also trap healthy blood cells, worsening the anemia. In some cases, doctors recommend removing the spleen to manage these effects.
Jaundice
Jaundice is the yellowing of the skin and the whites of the eyes. It occurs because hemolysis releases bilirubin, a yellow pigment produced when red blood cells break down. When bilirubin builds up faster than the liver can clear it, the telltale yellow tint appears.
For many people with alpha thalassemia HbH disease, jaundice comes and goes, often becoming more noticeable during illness or stress.
Bone Changes
Chronic anemia pushes the bone marrow to work harder, expanding in an attempt to produce more red blood cells. This expansion can reshape the bones over time. Facial features may change, with maxillary expansion (a prominent upper jaw) and frontal bossing (a protruding forehead).
Beyond facial changes, the disease can weaken bones. Osteoporosis—thinning, fragile bones—and bone pain are possible complications that need monitoring.
Growth and Development Issues
In children, chronic anemia can interfere with normal growth and development. Delayed growth and a later onset of puberty are recognized HbH disease signs and symptoms in younger patients. Because growing bodies demand more oxygen, the effects of anemia can be more visible during childhood. Early diagnosis allows doctors to monitor growth and step in when needed.
Gallstones
The ongoing breakdown of red blood cells doesn’t just cause jaundice—it also raises the risk of gallstones. Excess bilirubin can crystallize in the gallbladder, forming stones. These may cause pain or require treatment if they block the flow of bile.
Leg Ulcers
Less common, but still possible, leg ulcers can develop in people with Hemoglobin H disease. Poor oxygen delivery to the skin, especially around the ankles, can lead to slow-healing sores. While not everyone experiences them, they’re worth knowing about as part of the wider symptom picture.
Iron Overload
It may seem surprising that a condition causing anemia can also lead to too much iron. Yet iron overload is a real risk in HbH disease. It can happen for two reasons: the body absorbs more iron from food in response to anemia, and patients who receive frequent blood transfusions accumulate extra iron over time. Left unmanaged, iron overload can damage the heart, liver, and other organs.
Recognizing Early Signs and Symptoms
 Catching HbH disease signs and symptoms early can make a meaningful difference in how the condition is managed.
Catching HbH disease signs and symptoms early can make a meaningful difference in how the condition is managed.
When Do HbH Disease Signs and Symptoms Typically Appear?
Hemoglobin H disease is usually present from birth, but symptoms may not be obvious right away. In many cases, signs become noticeable during infancy or early childhood, as chronic anemia begins to affect energy, growth, and skin color. Some people have milder forms and aren’t diagnosed until later in life, often after a routine blood test reveals unusual results.
Subtle Indicators in Infancy and Childhood
In babies and young children, the early signs can be easy to miss. Look for:
- Persistent paleness
- Unusual fussiness or low energy
- Slower-than-expected growth
- Mild yellowing of the skin or eyes
These subtle clues, especially when combined, are worth discussing with a doctor.
Why Early Diagnosis Matters
Early diagnosis allows for proper monitoring and timely treatment. It helps doctors track growth in children, manage anemia before it causes complications, and watch for issues like iron overload. Catching the condition early also gives families the information they need for family planning decisions.
Differentiating from Mild Alpha-Thalassemia Symptoms
It’s important to separate HbH disease from milder forms of the condition. The trait and silent carrier states cause little or no symptoms, while HbH disease produces moderate, ongoing anemia. For a closer look at the gentler end of the spectrum, this guide on mild alpha thalassemia symptoms explains how the lighter forms behave. The clearest difference is severity—HbH disease symptoms are more persistent and more likely to need medical care.
Diagnosis of HbH Disease
A clear diagnosis combines clinical observation with targeted laboratory testing.
Clinical Suspicion Based on Symptoms
The diagnostic process often begins when a doctor notices signs of chronic anemia—fatigue, pallor, jaundice, or an enlarged spleen. A family history of thalassemia or ancestry from a high-prevalence region raises suspicion further. These clues prompt the laboratory tests that confirm the diagnosis.
Laboratory Tests
Several tests work together to confirm Hemoglobin H disease:
- Complete Blood Count (CBC): This first-line test typically reveals microcytic, hypochromic anemia—red blood cells that are smaller and paler than normal.
- Hemoglobin electrophoresis or HPLC: These methods separate and measure the different types of hemoglobin, allowing doctors to detect the presence of HbH directly.
- Genetic testing: DNA analysis is the definitive step, confirming the deletion of three alpha-globin genes.
For a complete walkthrough of the testing process, see this guide on thalassemia diagnosis methods. To understand what the numbers on your report mean, this thalassemia blood test guide breaks them down in detail.
Genetic testing is especially valuable because it confirms the exact gene changes and supports accurate family planning. According to the Centers for Disease Control and Prevention, proper testing also prevents thalassemia patients from being wrongly treated for iron deficiency.
Management and Treatment of HbH Disease Symptoms
While there’s no simple cure, Hemoglobin H disease can be managed effectively with the right care plan.
Regular Monitoring
Ongoing monitoring forms the foundation of care. Regular check-ups track hemoglobin levels, spleen size, growth in children, and signs of iron overload. This allows doctors to act before small problems become serious.
Folic Acid Supplementation
Because the body works hard to produce red blood cells, it needs plenty of folate. Folic acid supplements provide the building blocks for healthy red blood cell production and are a common, gentle part of management.
Blood Transfusions
Some people with alpha thalassemia HbH disease need blood transfusions, especially during periods of severe anemia—such as illness, surgery, or pregnancy. Transfusions are typically given as needed rather than on a constant schedule, depending on the individual’s symptoms.
Splenectomy
In selected cases where the spleen becomes severely enlarged or worsens the anemia, doctors may recommend removing it through a procedure called a splenectomy. This decision is carefully weighed, since the spleen plays a role in fighting infection.
Iron Chelation Therapy
If iron overload develops—whether from increased absorption or frequent transfusions—iron chelation therapy helps remove the excess. This treatment protects vital organs like the heart and liver from iron-related damage.
A Multidisciplinary Approach
Managing HbH disease often involves a team: hematologists, primary care doctors, and sometimes specialists in areas like bone health or cardiology. This coordinated approach ensures every aspect of the condition gets proper attention.
Lifestyle Modifications
Simple lifestyle steps support overall health. A balanced, folate-rich diet, avoiding unnecessary iron supplements, staying up to date on vaccinations, and keeping regular medical appointments all contribute to better outcomes.
Living with Hemoglobin H Disease
 A diagnosis of Hemoglobin H disease changes daily life, but with good care, most people manage well. Patient education is key—understanding your condition empowers you to recognize symptoms early, follow your treatment plan, and communicate effectively with your care team.
A diagnosis of Hemoglobin H disease changes daily life, but with good care, most people manage well. Patient education is key—understanding your condition empowers you to recognize symptoms early, follow your treatment plan, and communicate effectively with your care team.
Managing a chronic illness takes patience. Energy levels may fluctuate, and some days will feel harder than others. Building a strong support network, whether through family, friends, or patient communities, makes the journey easier.
The long-term outlook for people with HbH disease is generally positive, especially with consistent monitoring and care. Many lead full, active lives. The key is staying engaged with your healthcare team and addressing symptoms as they arise.
Conclusion
Hemoglobin H disease symptoms—chronic anemia, fatigue, jaundice, splenomegaly, and more—all trace back to a single genetic cause: three missing alpha-globin genes. Recognizing these signs early opens the door to proper diagnosis and effective management. With regular monitoring, supportive treatment, and a knowledgeable care team, people with alpha thalassemia HbH disease can manage the condition well and maintain a strong quality of life. Awareness, early diagnosis, and comprehensive care remain the most powerful tools for living well with this condition.
Frequently Asked Questions (FAQs)
1. What are the main HbH disease symptoms?
The main Hemoglobin H disease symptoms include chronic anemia, fatigue, weakness, pale skin, jaundice, and an enlarged spleen. Some people also develop gallstones, bone changes, or iron overload. Symptoms range from mild to moderate and are lifelong.
2. What causes Hemoglobin H disease?
HbH disease is caused by the deletion or malfunction of three of the four alpha-globin genes. With only one working gene, the body makes too little alpha-globin, leading to the formation of unstable Hemoglobin H and chronic anemia.
3. When do HbH disease signs and symptoms first appear?
Hemoglobin H disease is present from birth, and signs often appear in infancy or early childhood. Common early indicators include persistent paleness, low energy, slow growth, and mild jaundice. Milder cases may not be diagnosed until adulthood.
4. How is alpha thalassemia HbH disease diagnosed?
Diagnosis combines a Complete Blood Count (CBC), hemoglobin electrophoresis or HPLC, and genetic testing. The CBC shows small, pale red blood cells, electrophoresis detects HbH, and genetic testing confirms the deletion of three alpha-globin genes.
5. Is HbH disease the same as the alpha thalassemia trait?
No. The alpha thalassemia trait involves two missing genes and causes little or no symptoms. HbH disease involves three missing genes and causes moderate, ongoing anemia. HbH disease is more severe and usually requires medical care.
6. Can Hemoglobin H disease be cured?
There is no simple cure for HbH disease, but it can be managed effectively. Treatment includes regular monitoring, folic acid, blood transfusions when needed, and iron chelation therapy if iron overload develops. Most people live full, active lives.
7. Why does HbH disease cause iron overload despite anemia?
Iron overload can occur because the body absorbs more iron from food in response to anemia, and frequent blood transfusions add extra iron. Over time, this excess can harm organs, which is why iron levels are monitored closely.
8. Do all people with HbH disease need blood transfusions?
No. Many people with alpha thalassemia HbH disease only need transfusions occasionally—during illness, surgery, pregnancy, or periods of severe anemia. The need depends on each person’s symptoms and hemoglobin levels.
9. Is HbH disease hereditary?
Yes. Hemoglobin H disease is inherited. It develops when a child inherits alpha-globin gene deletions from both parents. This is why genetic counseling and carrier screening matter for couples planning a family.
10. What is the long-term outlook for someone with HbH disease?
The outlook is generally positive. With consistent monitoring, supportive treatment, and a strong care team, most people with HbH disease manage their symptoms well and lead active lives. Early diagnosis and ongoing care are key to good outcomes.