


People with HbH disease can achieve a good long-term outlook with early diagnosis, regular monitoring, and appropriate treatment. Managing complications such as anemia, iron overload, and organ damage through ongoing medical care helps improve quality of life and supports better long-term health outcomes.
HbH disease complications span multiple body systems, including chronic anemia, splenomegaly, iron overload, cardiovascular issues, endocrine disruption, and neurological events. These arise from three missing alpha-globin genes that impair hemoglobin production. Early diagnosis, regular monitoring, and targeted HbH disease treatment significantly reduce long-term risks and improve quality of life.
Hemoglobin H disease sits in a complicated middle ground. It’s more serious than the alpha thalassemia trait, yet far less severe than the fatal hydrops fetalis form. What makes HbH disease particularly challenging is its reach—alpha thalassemia complications don’t stop at the blood. They extend into the heart, liver, bones, endocrine system, reproductive organs, and beyond.
Understanding HbH disease complications matters because the condition is lifelong. People living with Hemoglobin H disease symptoms from childhood face decades of managing not just anemia, but the cascading consequences of chronic red blood cell destruction and iron accumulation. The earlier these complications are identified, the better the chances of preventing serious organ damage.
This guide covers the full spectrum of HbH disease complications—from hematological and cardiovascular issues to endocrine, neurological, and reproductive concerns. You’ll also find a breakdown of how each complication is monitored, how HbH disease treatment addresses them, and what the future of management looks like. For a foundational overview of the condition itself, this guide on HbH disease symptoms explained provides essential context.
What Causes HbH Disease Complications?
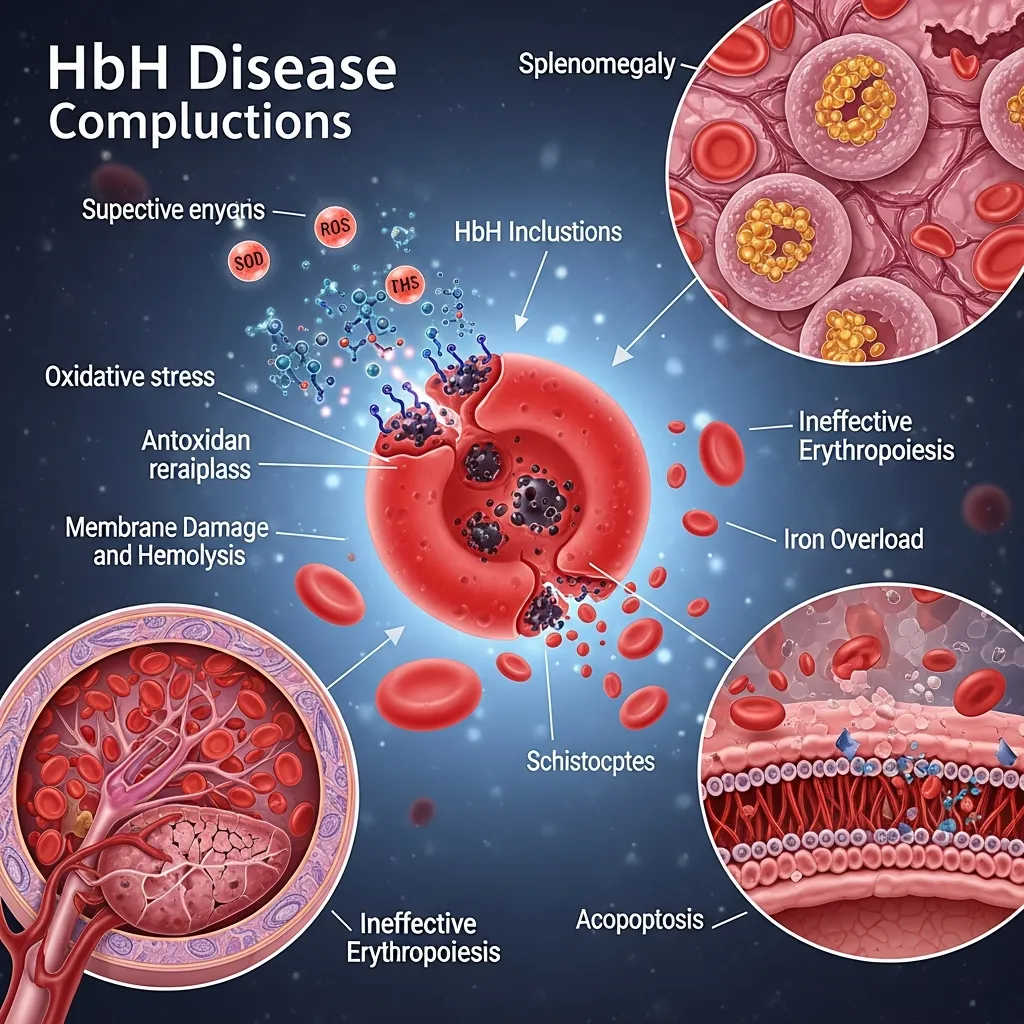 HbH disease develops when three of the four alpha-globin genes are deleted or non-functional. With just one working gene, the body produces far too little alpha-globin. Excess beta-globin chains clump together to form Hemoglobin H—an unstable molecule that damages red blood cells from within and breaks them down rapidly.
HbH disease develops when three of the four alpha-globin genes are deleted or non-functional. With just one working gene, the body produces far too little alpha-globin. Excess beta-globin chains clump together to form Hemoglobin H—an unstable molecule that damages red blood cells from within and breaks them down rapidly.
This process, called hemolysis, triggers chronic anemia and forces the bone marrow to work harder. Over time, the consequences ripple outward. Iron accumulates. The spleen enlarges. Organs that depend on steady oxygen delivery—the heart, liver, endocrine glands—begin to suffer. The result is a multi-system disease that demands comprehensive, ongoing care.
To understand how HbH disease differs from milder forms of the alpha thalassemia spectrum, see this detailed resource on mild alpha thalassemia symptoms.
Hematological Complications of HbH Disease
How does chronic anemia affect people with HbH disease?
Chronic anemia is the hallmark HbH disease complication. Hemoglobin levels are persistently low, typically ranging between 7 and 10 g/dL. Fatigue, pallor, shortness of breath, and reduced exercise tolerance are the everyday consequences.
Two processes drive this anemia simultaneously: ineffective erythropoiesis (the bone marrow producing faulty red blood cells) and accelerated hemolysis (those cells breaking down far too quickly). Neither compensatory mechanism fully corrects the deficiency, leaving the body in a constant state of oxygen shortage.
What is splenomegaly and hypersplenism in HbH disease?
The spleen acts as a filter, clearing damaged red blood cells. In Hemoglobin H disease, it operates under relentless pressure. The constant influx of fragile, abnormal cells causes progressive splenic enlargement—splenomegaly—which can become severe enough to cause abdominal discomfort and a feeling of fullness.
Hypersplenism follows when an overactive spleen begins trapping not just damaged cells but healthy ones too. This worsens anemia and reduces platelet and white blood cell counts, increasing the risk of bleeding and infection.
Does HbH disease cause iron overload?
Iron overload is a paradoxical but serious alpha thalassemia complication. Despite chronic anemia, the body absorbs more iron from food as compensation. Patients who receive regular blood transfusions as part of their HbH disease treatment accumulate additional iron over time.
Without chelation therapy, excess iron deposits in the liver, heart, and endocrine organs, causing progressive damage. Left unmanaged, iron overload becomes one of the most dangerous long-term complications of Hemoglobin H disease.
What is erythroid hyperplasia, and how does it affect bones?
When the bone marrow expands to produce more red blood cells, it remodels the surrounding bone. This erythroid hyperplasia can cause bone thinning, osteoporosis, and structural changes—particularly in the skull and face. A prominent forehead (frontal bossing) and expanded upper jaw (maxillary expansion) are recognized Hemoglobin H disease symptoms in children with inadequately managed anemia.
Cardiovascular Complications
Can HbH disease cause pulmonary hypertension?
Chronic hemolysis releases free hemoglobin into the bloodstream. This scavenges nitric oxide—a molecule critical for keeping blood vessels relaxed and dilated. The result is progressive vasoconstriction in the pulmonary vessels, raising pressure in the lungs and increasing the risk of pulmonary hypertension.
According to research in thalassemia populations, pulmonary hypertension significantly elevates the risk of heart failure and premature death. Regular echocardiographic screening is essential for monitoring this complication.
What is cardiac iron overload and cardiomyopathy in HbH disease?
Excess iron that accumulates in the heart muscle disrupts electrical conduction and impairs contractility. Over time, cardiac iron overload leads to cardiomyopathy—a weakening of the heart muscle that can cause arrhythmias, heart failure, and sudden cardiac death. This remains one of the leading causes of mortality in transfusion-dependent thalassemia patients.
Cardiac MRI is the most accurate tool for quantifying myocardial iron and guiding chelation decisions.
What is the risk of thromboembolic events in HbH disease?
Hemoglobin H disease increases the risk of blood clots. Chronic hemolysis releases cellular components that activate clotting pathways, while splenectomy—performed in some patients—further elevates this risk by removing the organ responsible for clearing activated platelets and coagulation factors. Deep vein thrombosis, pulmonary embolism, and stroke have all been documented in HbH patients, particularly those who have undergone splenectomy.
Endocrine Complications
How does HbH disease affect growth and puberty?
Chronic anemia and iron deposition in the pituitary gland disrupt growth hormone secretion and the hormonal signals that trigger puberty. Children with Hemoglobin H disease may experience delayed linear growth and a later onset of secondary sexual development. Monitoring growth velocity and hormonal markers from an early age allows timely intervention with growth hormone therapy or hormone replacement.
What endocrine organs does iron overload damage in HbH disease?
Iron deposits accumulate in multiple glands, causing:
- Hypothyroidism: Iron in thyroid tissue impairs hormone production, leading to fatigue, weight gain, cold intolerance, and cognitive slowing.
- Diabetes mellitus: Pancreatic iron overload damages insulin-producing beta cells, causing secondary diabetes that differs from type 1 or type 2 diabetes and requires careful management.
- Hypoparathyroidism: Iron damage to the parathyroid glands disrupts calcium regulation, causing muscle cramps, tingling, and in severe cases, tetany.
Each of these endocrine complications reinforces the urgency of effective iron chelation as a core component of HbH disease treatment.
Hepatic and Gallbladder Complications
What liver complications develop in HbH disease?
The liver is the primary storage site for excess iron. As iron accumulates, hepatocytes sustain oxidative damage, triggering inflammation and progressive fibrosis. Without effective chelation, this can advance to cirrhosis—irreversible scarring that impairs liver function, raises the risk of liver failure, and increases vulnerability to hepatocellular carcinoma.
Regular liver iron concentration measurements (via MRI or liver biopsy) and liver function tests are standard components of HbH disease monitoring protocols.
Why does HbH disease increase the risk of gallstones?
Chronic hemolysis liberates large amounts of bilirubin—the yellow pigment produced when red blood cells break down. When bilirubin overwhelms the liver’s capacity to process it, the excess crystallizes in the gallbladder, forming pigment gallstones (cholelithiasis).
Gallstones can be asymptomatic or cause recurrent episodes of biliary colic, cholecystitis, and bile duct obstruction. Ultrasound surveillance is recommended in patients with ongoing hemolysis, and cholecystectomy may be necessary when stones become symptomatic.
Infectious Complications
Why are people with HbH disease more susceptible to infections?
Several overlapping factors increase infection risk in HbH disease. Splenomegaly impairs the spleen’s ability to efficiently filter bacteria from the bloodstream. Hypersplenism reduces white blood cell counts. Iron overload, paradoxically, promotes the growth of iron-dependent pathogens such as Yersinia enterocolitica and certain fungal organisms.
How does splenectomy increase the risk of infection in HbH disease?
Splenectomy resolves hypersplenism but eliminates the spleen’s protective functions entirely. Post-splenectomy patients face a lifelong elevated risk of overwhelming sepsis from encapsulated bacteria, particularly Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis. According to the Centers for Disease Control and Prevention, individuals without a spleen require specific vaccinations and sometimes prophylactic antibiotics to reduce this risk. This decision must be weighed carefully before proceeding with splenectomy in HbH disease management.
Neurological Complications
What neurological complications can occur in HbH disease?
Stroke and transient ischemic attacks (TIAs) are recognized complications, particularly in patients with pulmonary hypertension, thromboembolic risk factors, or post-splenectomy hypercoagulability. Cerebrovascular events can cause permanent neurological deficits if not rapidly treated.
Rare but documented cases of spinal cord compression have also been reported, caused by extramedullary hematopoiesis—when the expanding bone marrow spills into spaces outside the bone and presses on neural structures. This may present as back pain, limb weakness, or sensory changes and requires urgent imaging and treatment.
Reproductive Complications
How does HbH disease affect fertility and pregnancy?
Iron overload in the pituitary and gonads disrupts the hormonal signaling required for ovulation and sperm production. Males may develop hypogonadism with impaired sperm quality; females may experience irregular or absent menstrual cycles and difficulty conceiving.
Pregnancy in HbH disease carries additional risks, including worsening anemia, increased transfusion requirements, preterm delivery, and fetal growth restriction. Management requires close collaboration between hematologists, obstetricians, and maternal-fetal medicine specialists throughout the pregnancy. For information on how alpha thalassemia genetics affect family planning decisions, this guide on alpha thalassemia trait symptoms provides useful context.
Psychological and Quality of Life Issues
How does living with HbH disease affect mental health and daily life?
A chronic, lifelong blood disorder imposes a psychological burden that rarely receives the same attention as physical complications. Fatigue, frequent medical appointments, hospitalizations, and uncertainty about disease progression contribute to higher rates of anxiety and depression among HbH disease patients. The World Health Organization recognizes the psychosocial dimensions of chronic illness as essential components of comprehensive care.
Economic pressures compound these difficulties. Treatment costs, time away from work or school, and reliance on others for support create social and financial strain. Patient education—giving individuals a clear understanding of their condition and its management—significantly improves adherence to treatment and overall quality of life. Support groups and peer networks provide emotional validation and practical strategies for living well with the disease.
Diagnosis and Monitoring of HbH Disease Complications
Effective management of HbH disease complications depends on systematic, proactive screening.
Standard monitoring protocols include:
- Regular complete blood counts (CBC) to track hemoglobin levels and blood cell indices
- Serum ferritin and transferrin saturation to assess iron burden
- Liver MRI (T2)* for accurate liver iron concentration measurement
- Cardiac MRI (T2)* to detect early myocardial iron deposition
- Echocardiography for pulmonary hypertension screening
- Endocrine panel including thyroid function, glucose, calcium, and pituitary hormones
- Bone density (DEXA scan) for osteoporosis surveillance
- Liver function tests and abdominal ultrasound for hepatic and gallbladder assessment
Genetic testing remains the gold standard for confirming the diagnosis of HbH disease and supporting family planning decisions. For a complete guide to the diagnostic process, see this resource on thalassemia diagnosis methods.
Management and Treatment of HbH Disease Complications
What is the role of blood transfusions and chelation therapy in HbH disease treatment?
Blood transfusions manage severe or symptomatic anemia, particularly during illness, surgery, or pregnancy. Unlike transfusion-dependent thalassemia major, most HbH patients require transfusions only episodically. Each transfusion, however, contributes to iron accumulation.
Iron chelation therapy—using agents such as deferoxamine, deferasirox, or deferiprone—removes excess iron and protects the heart, liver, and endocrine organs. Regular iron monitoring guides chelation dose adjustments to avoid both under-treatment and over-chelation.
When is splenectomy indicated in HbH disease?
Splenectomy is considered when splenomegaly becomes severe, when hypersplenism substantially worsens anemia or reduces platelet/white blood cell counts, or when the spleen causes significant discomfort. The decision is never taken lightly, given the lifelong infection risk it creates. Vaccination and, in some cases, prophylactic penicillin are mandatory following splenectomy.
What emerging treatments are available for HbH disease?
Gene therapy represents the most promising frontier in HbH disease treatment. Techniques that restore alpha-globin gene function—or that use gene editing to increase fetal hemoglobin production as a compensatory strategy—have shown strong results in clinical trials for related hemoglobinopathies. Luspatercept, an erythroid maturation agent, is being investigated for its potential to reduce transfusion burden in thalassemia patients. Bone marrow transplantation offers a curative option in selected severe cases, though donor availability and transplant-related risks limit its broader application.
What Is the Long-Term Outlook for People with HbH Disease?

How can people with HbH disease improve their quality of life?
The long-term outlook for Hemoglobin H disease has improved substantially with advances in monitoring and treatment. Most patients who receive consistent, comprehensive care live into adulthood and beyond. Staying engaged with a multidisciplinary care team—including hematologists, cardiologists, endocrinologists, and mental health professionals—remains the single most effective strategy for preventing serious organ damage.
Patient education, genetic counseling for family planning, and access to peer support communities all play meaningful roles in helping people with HbH disease lead full, active lives.
Conclusion
HbH disease complications are broad, persistent, and interconnected—but they are manageable. The key lies in treating the condition as the multi-system disease it is, rather than focusing on anemia alone. Cardiovascular, endocrine, hepatic, neurological, reproductive, and psychological dimensions all demand attention alongside the core hematological issues.
Advances in cardiac MRI monitoring, newer chelation agents, and emerging gene therapies are already reshaping the prognosis for patients born with this condition today. The next decade holds genuine promise for curative approaches. Until then, comprehensive monitoring, timely HbH disease treatment, and patient education remain the most powerful tools available.
If you or someone you know is living with Hemoglobin H disease, work closely with a multidisciplinary team, stay consistent with monitoring appointments, and connect with patient communities for support and shared experience.
Frequently Asked Questions About HbH Disease Complications
1. What are the most serious HbH disease complications to watch for?
The most serious HbH disease complications include cardiac iron overload and cardiomyopathy, liver fibrosis and cirrhosis, pulmonary hypertension, thromboembolic events, and endocrine failure. These develop gradually and are best detected through regular, proactive monitoring rather than waiting for symptoms to appear.
2. Can HbH disease complications be prevented?
Many alpha thalassemia complications can be significantly delayed or reduced in severity with early diagnosis and consistent treatment. Iron chelation therapy protects organs from iron-related damage, and routine screening catches complications before they become irreversible.
3. Does everyone with HbH disease develop iron overload?
Not necessarily. Iron overload risk depends on transfusion frequency and the body’s baseline iron absorption. Patients who rarely need transfusions may develop iron overload more slowly. Regular monitoring with ferritin levels and imaging ensures chelation is started at the right time.
4. How does HbH disease affect children differently from adults?
In children, HbH disease complications particularly affect growth, bone development, and puberty. Inadequately managed anemia during childhood can cause bone deformities, delayed growth, and late puberty. Adult patients face cumulative iron accumulation and its associated cardiac, hepatic, and endocrine effects.
5. What is the connection between HbH disease and pulmonary hypertension?
Chronic hemolysis in HbH disease depletes nitric oxide, causing pulmonary vascular constriction and progressive pulmonary hypertension. This is a serious complication that can lead to right heart failure. Echocardiographic screening is recommended for all HbH patients.
6. Is splenectomy always recommended for enlarged spleen in HbH disease?
No. Splenectomy is reserved for cases where splenomegaly causes severe hypersplenism or significant symptoms. The procedure carries a lifelong risk of serious infections from encapsulated bacteria, so the decision requires careful risk-benefit assessment and mandatory vaccinations.
7. How does HbH disease affect fertility and pregnancy outcomes?
Iron overload in the pituitary and gonads can impair fertility in both males and females. Pregnancy in HbH disease carries risks of worsening anemia, preterm delivery, and fetal growth restriction, requiring specialist co-management throughout.
8. What medications are used in HbH disease treatment for iron overload?
The three main iron chelators used in HbH disease treatment are deferoxamine (given by infusion), deferasirox (oral tablet or dispersible tablet), and deferiprone (oral tablet). The choice depends on the patient’s iron burden, organ function, and tolerability.
9. Are there any curative treatments for HbH disease?
Bone marrow transplantation can cure Hemoglobin H disease in selected patients, but it carries significant risks and requires a suitable donor. Gene therapy approaches are showing strong promise in clinical trials and may offer a safer curative option in the future.
10. How is HbH disease different from other forms of alpha thalassemia?
HbH disease results from three missing alpha-globin genes and causes moderate-to-significant chronic anemia requiring active management. The alpha thalassemia trait (two missing genes) and silent carrier state (one missing gene) cause little or no symptoms and rarely need treatment. HbH disease sits a step below the most severe form, hydrops fetalis, which involves all four missing genes and is typically fatal.