


Hemoglobin H is a form of alpha thalassemia that results from reduced alpha-globin production, leading to abnormal hemoglobin formation. It causes chronic anemia, fatigue, and enlarged spleen, and is managed through ongoing medical care and monitoring.
The Genetic Basis of Hemoglobin H Disease
To understand this condition, you have to look at your DNA. Human cells rely on a precise set of genetic instructions to build healthy red blood cells.
Alpha-globin genes and their role
Normal adult hemoglobin is made of four protein chains: two alpha chains and two beta chains. The production of the alpha chains is controlled by four specific genes. You inherit two of these alpha-globin genes from your mother and two from your father. When all four genes function properly, the body produces a perfectly balanced amount of hemoglobin to transport oxygen efficiently.
How deletions lead to HbH disease
Hemoglobin h disease occurs when three of these four alpha-globin genes are missing or non-functional. With only one working alpha gene, the body simply cannot produce enough alpha-globin chains. This severe shortage disrupts the normal pairing of alpha and beta chains, leading to structurally unstable hemoglobin.
Understanding the inheritance patterns
Because it is a genetic condition, it is passed down through families. A child develops this disease if they inherit two missing genes from one parent (a trait carrier) and one missing gene from the other parent (a silent carrier). Understanding this specific inheritance pattern is critical for family planning and assessing the risk for future generations.
Pathophysiology: How Hemoglobin H Affects the Body
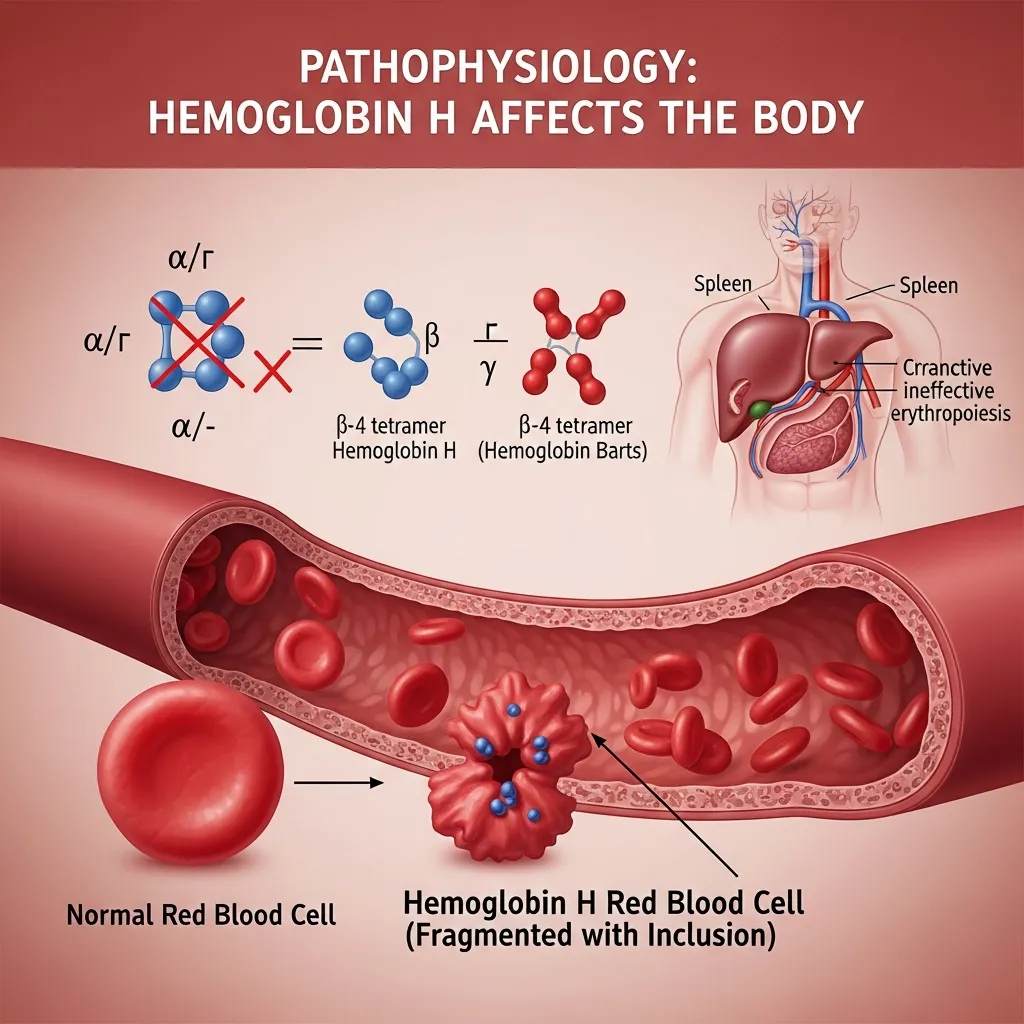 The missing genes trigger a chain reaction inside the body, fundamentally changing how red blood cells operate and survive.
The missing genes trigger a chain reaction inside the body, fundamentally changing how red blood cells operate and survive.
The formation of beta-globin tetramers
Because there is a severe lack of alpha chains, the beta chains have no partners. Left without alpha chains to bind to, these excess beta chains group together in clusters of four. These abnormal clusters are called beta-globin tetramers, or “Hemoglobin H.”
Hemoglobin H and oxidative stress
While these tetramers can carry oxygen, they are entirely ineffective at delivering it to the body’s tissues. Furthermore, they are highly unstable. As they break down inside the red blood cells, they create oxidative stress. This stress damages the cell’s internal structures and its outer membrane.
Red blood cell damage and hemolytic anemia
The damaged red blood cells become rigid and deformed. As they pass through the spleen—the body’s blood-filtering organ—they are recognized as abnormal and destroyed prematurely. This rapid destruction of red blood cells is known as hemolytic anemia, which forces the bone marrow to work overtime to replace the lost cells.
Clinical Manifestations and Symptoms
The symptoms of hemoglobin h disease vary widely from person to person. Some individuals experience mild complications, while others face more severe daily challenges.
Anemia and its severity
Chronic anemia is the most common symptom. Because red blood cells are destroyed faster than they can be replaced, patients often experience fatigue, weakness, and a pale complexion. The severity of the anemia can fluctuate, often worsening during periods of illness, infection, or pregnancy.
Splenomegaly and its complications
The spleen works constantly to filter out the damaged red blood cells. This heavy workload causes the spleen to enlarge, a condition known as splenomegaly. An enlarged spleen can cause abdominal discomfort, a feeling of fullness after eating very little, and an increased risk of spleen rupture during physical impact.
Jaundice, gallstones, and other associated symptoms
When red blood cells break down, they release a yellow pigment called bilirubin. High levels of bilirubin cause jaundice, a yellowing of the skin and the whites of the eyes. Over time, the excess bilirubin can clump together in the gallbladder, forming painful gallstones that may require surgical removal.
Skeletal changes and growth retardation
To compensate for the rapid loss of blood cells, the bone marrow expands to produce more. This expansion can cause skeletal changes, particularly in the facial bones and skull. In children, the immense energy required to constantly produce new blood cells can sometimes lead to delayed growth and delayed puberty.
Diagnosing Hemoglobin H Disease
Accurate diagnosis prevents unnecessary treatments and ensures patients receive the right supportive care.
Initial screening tests
The diagnostic process typically starts with a Complete Blood Count (CBC) and a blood film examination. Patients usually show a low red blood cell count, and the cells themselves appear abnormally small and pale under a microscope.
Hemoglobin electrophoresis and HPLC
To confirm the type of hemoglobin present, doctors use a test called hemoglobin electrophoresis or High-Performance Liquid Chromatography (HPLC). These tests separate the different types of hemoglobin in the blood. The presence of the fast-moving Hemoglobin H tetramers strongly indicates the disease.
Genetic testing for definitive diagnosis
For absolute certainty, targeted genetic testing is utilized. DNA analysis maps the alpha-globin genes to pinpoint exactly how many are missing. This removes all guesswork and provides a definitive diagnosis.
Prenatal diagnosis methods
If both parents are known carriers of alpha thalassemia traits, prenatal testing can determine the genetic status of the fetus. Procedures like chorionic villus sampling (CVS) or amniocentesis are performed early in the pregnancy to provide parents with vital information.
Management and Treatment Strategies
 While there is no universal cure, comprehensive medical management helps patients lead active, fulfilling lives.
While there is no universal cure, comprehensive medical management helps patients lead active, fulfilling lives.
Regular monitoring and supportive care
Consistent check-ups with a hematologist form the foundation of care. Doctors routinely monitor hemoglobin levels, organ function, and growth in children to catch any complications early.
Blood transfusions: indications and risks
Many individuals with hemoglobin h disease only need occasional blood transfusions during times of severe stress, such as a major infection or surgery. However, some patients experience more severe anemia and require regular transfusions to maintain healthy oxygen levels.
Iron chelation therapy: preventing iron overload
A major complication of frequent transfusions—and the disease itself—is the buildup of excess iron in the body. Since the body has no natural way to excrete extra iron, patients often require iron chelation therapy. These medications bind to excess iron and remove it through urine or stool, protecting the heart and liver from damage.
Splenectomy: considerations and outcomes
If the spleen becomes massively enlarged or starts destroying healthy red blood cells alongside the damaged ones, a surgeon may recommend a splenectomy (removal of the spleen). While this can improve hemoglobin levels, it also increases the lifelong risk of severe infections.
Folic acid supplementation
Because the bone marrow is constantly producing new red blood cells, the body burns through its stores of folic acid rapidly. Doctors frequently prescribe daily folic acid supplements to support healthy blood cell production.
Living with Hemoglobin H Disease: Daily Care and Lifestyle
Medical treatments are only part of the equation. Daily habits play a massive role in maintaining energy and overall well-being.
Dietary considerations and nutrition
Eating a balanced diet supports general health, but patients must be mindful of their iron intake. Avoiding iron supplements and limiting highly iron-fortified foods helps prevent iron overload. Drinking black tea with meals can also reduce the amount of iron the digestive system absorbs.
Importance of regular exercise
Moderate, low-impact exercise like walking, swimming, or cycling improves cardiovascular health and boosts mood. Patients should listen to their bodies and avoid extreme exertion, especially on days when anemia symptoms feel more pronounced.
Managing fatigue and improving quality of life
Pacing daily activities prevents severe exhaustion. Breaking large tasks into smaller steps and prioritizing rest ensures that energy levels remain stable throughout the day.
Psychological support and coping mechanisms
Living with a chronic genetic disorder takes an emotional toll. Joining support groups, seeking counseling, and maintaining open communication with friends and family helps patients manage the mental health challenges associated with their diagnosis.
Emerging Therapies and Future Directions
The landscape of genetic blood disorders is shifting rapidly, bringing new hope to patients globally.
Gene therapy and gene editing
Researchers are actively exploring gene-editing technologies like CRISPR to correct the underlying genetic defects. By repairing or replacing the missing alpha-globin genes at the cellular level, scientists hope to offer a permanent cure in the future.
New drug developments
Pharmaceutical companies are developing new drugs aimed at improving red blood cell maturation and reducing the destruction of cells in the spleen. These medications could eventually reduce the need for blood transfusions.
Bone marrow transplantation
Currently, a bone marrow transplant (or stem cell transplant) is the only potential cure. It replaces the defective blood-forming stem cells with healthy ones from a compatible donor. Because the procedure carries significant risks, it is usually reserved for the most severe cases.
Complications of Hemoglobin H Disease
Proactive management is necessary to prevent long-term complications affecting various organ systems.
Iron overload and organ damage
Even without regular transfusions, the body naturally absorbs more iron from food to compensate for the chronic anemia. Over time, this excess iron accumulates in the heart, liver, and endocrine glands, leading to organ failure if left untreated.
Bone complications and osteoporosis
The expansion of the bone marrow weakens the outer layers of the bone. This increases the risk of fractures and early-onset osteoporosis. Adequate calcium and vitamin D intake, along with regular bone density scans, are essential preventive measures.
Infections and immune compromise
Patients who have had their spleen removed face a significantly higher risk of bacterial infections. Staying up to date with vaccines, including the pneumococcal and flu vaccines, is mandatory. The Centers for Disease Control and Prevention (CDC) provides strict vaccination guidelines for immunocompromised individuals.
Cardiovascular complications
Chronic anemia forces the heart to pump harder to deliver sufficient oxygen to the body. Over many years, this increased workload can lead to an enlarged heart, arrhythmias, or heart failure.
Differentiation from Other Thalassemia Syndromes
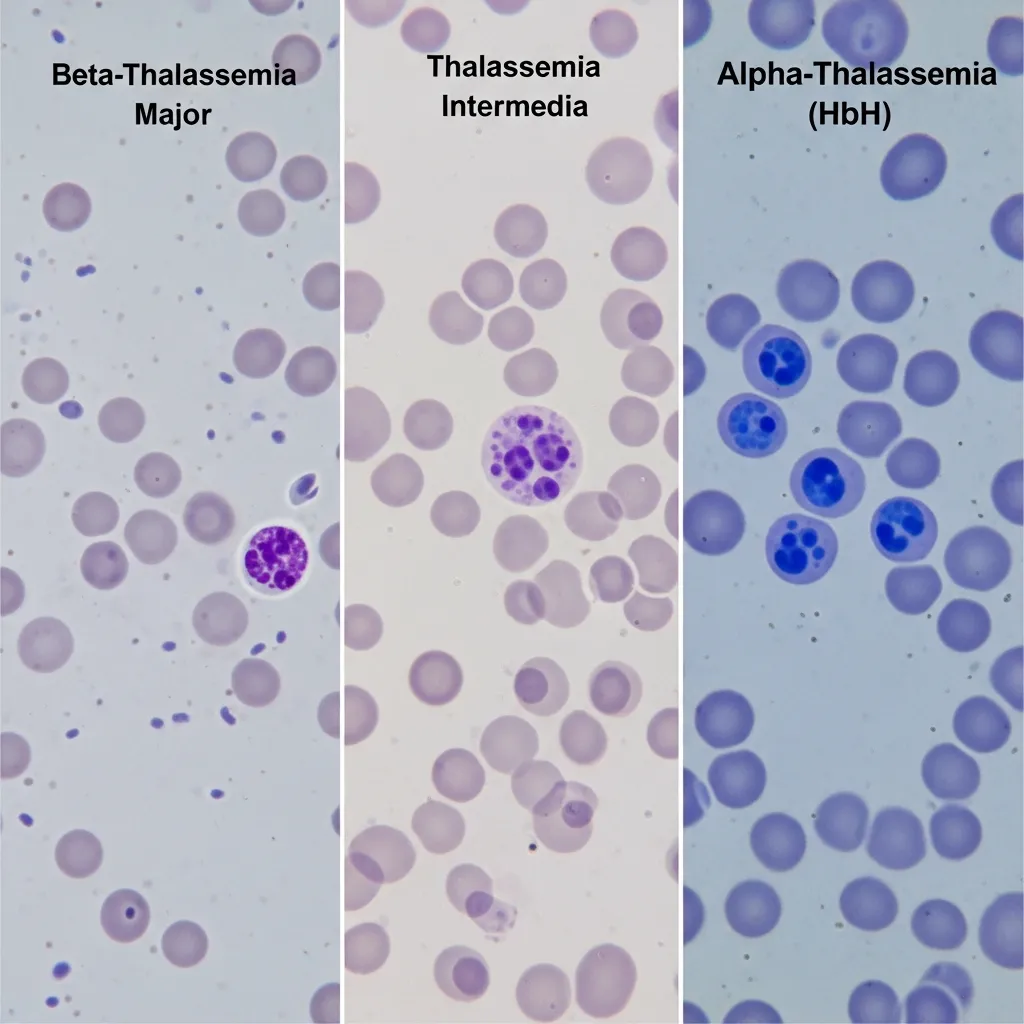 Because symptoms overlap, doctors must carefully distinguish hemoglobin h disease from related conditions.
Because symptoms overlap, doctors must carefully distinguish hemoglobin h disease from related conditions.
Alpha thalassemia silent carrier
A silent carrier is missing only one alpha-globin gene. They have no symptoms, perfectly normal hemoglobin levels, and require no treatment.
Alpha thalassemia trait
Also known as alpha thalassemia minor, this occurs when two genes are missing. It causes very mild anemia and slightly smaller red blood cells, but patients generally live completely normal lives without medical intervention.
Hydrops fetalis (Barts Hydrops)
This is the most severe form of alpha thalassemia, occurring when all four alpha-globin genes are missing. It is almost always fatal before or shortly after birth, as the fetus cannot produce any functional hemoglobin.
Beta thalassemia major and intermedia
While alpha thalassemia affects the alpha chains, beta thalassemia affects the beta chains. Beta thalassemia major requires lifelong, frequent blood transfusions starting in early childhood, making it generally more severe than the average case of hemoglobin h disease.
The Role of Genetic Counseling
Understanding your genetics is a powerful tool for planning your family’s future.
Importance for affected individuals and carriers
Genetic counselors help patients interpret their DNA test results. They explain exactly what it means to carry these genetic deletions and how it impacts personal health.
Family planning and reproductive options
If a patient wishes to have children, the counselor will strongly recommend testing the partner. Knowing both parents’ genetic status allows the counselor to calculate the exact statistical risk of passing the condition to the child. They also guide parents through options like prenatal screening or in vitro fertilization (IVF) with genetic selection.
Frequently Asked Questions (FAQs)
1. What is Hemoglobin H disease?
Hemoglobin H disease is a type of alpha thalassemia where the body produces too little alpha-globin, leading to abnormal hemoglobin and chronic anemia.
2. What causes Hemoglobin H disease?
It is caused by genetic mutations in the alpha-globin genes, usually inherited from both parents.
3. Is Hemoglobin H disease inherited?
Yes, it is an inherited blood disorder passed down through families.
4. What are the common symptoms of Hemoglobin H disease?
Common symptoms include fatigue, weakness, pale skin, and an enlarged spleen.
5. How is Hemoglobin H disease diagnosed?
It is diagnosed through blood tests, hemoglobin analysis, and genetic testing.
6. Is Hemoglobin H disease a severe condition?
It can range from mild to moderate severity, but some patients may require regular treatment.
7. Can Hemoglobin H disease be cured?
There is no complete cure, but symptoms can be managed with proper medical care.
8. What treatments are available for Hemoglobin H disease?
Treatment may include blood transfusions, folic acid supplements, and regular monitoring.
9. Does Hemoglobin H disease affect daily life?
Yes, in some cases it can cause fatigue and reduced physical endurance, but many people live normal lives with care.
10. Can Hemoglobin H disease be prevented?
It cannot be prevented, but genetic counseling can help identify risks before having children.
Conclusion
Hemoglobin H disease is a genetic blood disorder that affects hemoglobin production and leads to chronic anemia. Although it has no permanent cure, early diagnosis and proper medical management can significantly improve quality of life. Regular monitoring, supportive treatment, and genetic awareness are key to managing this condition effectively.